
一般认为,当一个除菌级过滤器的非破坏性物理完整性试验(例如:前进流扩散试验或者泡点试验)与细菌截留数据相关联,且重复试验仍然可以证明其完整性,那么此重复使用过滤器是可靠的并能提供无菌的滤出液。前进流和泡点试验的方法都能够高效检测过滤器生产中的缺点与不足,例如:膜的大孔或者由于安装不当而产生的滤芯密封旁路。在不兼容的重复使用条件下,膜有可能发生降解并危及到过滤器的细菌截留性质,但这种情况往往被忽视。过滤器厂商的核心验证研究没有对这种膜老化的情况进行模拟;使用在通常模型状态下与截留相关联的普通的产品完整性试验根本无法检测膜老化过程。
过滤器重复使用的法规指南
和ICH(人用药物注册技术要求国际协调会议)标准
回顾美国食品和药物管理局(FDA)和欧洲药品局(EMEA)有关API和非无菌药物的药品生产管理规范表明,对非无菌APIs或者非无菌的最终药物产品,无论除菌级过滤器的使用或重复使用都没有特别涉及到。该指南也针对非无菌API和生物技术APIs从发酵到下游分离纯化生产过程中除菌级过滤器的使用,包括无菌过滤培养基、添加剂、缓冲液。虽然在非无菌API和非无菌药物生产过程使用除菌级过滤器会考虑现行药品生产管理规范,但是关于除菌级过滤器的使用和重复使用的特殊指导仅由无菌药物指南提出。

图1 没有进行全重复使用工艺规模的验证,就重复使用除菌级过滤器具有一定的风险。
药物活性成分生产管理规范指南中有这样说法:非无菌API可以考虑除菌级过滤器的重复使用。“构建的设备其与原材料、中间体或API接触的表面不得对中间体或API品质有超过正式或其它已公布的规格以外的影响。”根据该指南,除了除菌级过滤器对API的潜在影响,包括过滤器厂商可能提供的溶出物数据以外,当在API生产中重复使用过滤器时,终端用户也应该考虑其他对产品的潜在影响,如:截留的细菌,未充分去除的产品及过滤器上清洗剂的残留。对于无菌药物生产过程,cGMP的相关规定如下:“构建的设备其与成分、工艺中材料或药物产品接触的表面不得有反应、添加或吸附从而对药品的安全性、特性、强度、质量或纯度产生超过正式或其他已公布的要求以外的影响。”
无菌过程生产无菌药物产品的工业规范对于重复使用的过滤器表面的潜在影响有着同样的考虑。现行GMP对除菌过滤器的使用和重复使用过程提出了两点看法。关于无菌药物产品规定:“单一批次生产之后的除菌过滤器应该进行常规的报废处理。”虽然这条规定看似不支持除菌过滤器的重复使用,但是接下来指南陈述道:“然而,在重复使用能被评估的情形下,除菌过滤器的验证应当包括将处理的最大数量批次。”这一通融条款说明:重复使用除菌级过滤器时,除菌过滤器工艺验证必须评估终端用户工艺中清洁、再灭菌过程的影响。规范还提到:“重要的是过滤后完整性检测能够进行以发现可能在过滤过程中发生的任何过滤器泄漏或穿孔。”这个法规同时适用于分隔的多批次和多批次组和。关键点在于,规范指出过滤器可能危及除菌性能的泄露和穿孔将以完整性试验来检测。
欧洲指南
欧委会关于人用和兽用药品生产管理规范中提到:“在没有经过验证的情况下,同一个过滤器最多只能使用1个工作日。”这句话似乎允许延长使用时间和可能的重复使用,但是同时过程验证应该考虑到重复使用对过滤器产生的任何影响。如API生产规范中所提的一样,规范中还指出“过滤器不得影响产品,不论是去除组分或释出物质。”这里再次提到过滤器重复使用前,必须考虑使用过的过滤器上污染物或清洗剂的析出,任何类似的影响必须验证。
PDA的建议
关于除菌级过滤器重复使用的行业建议已经受到时间局限。1988年注射用药物协会(PDA)第一版26号技术报告“液体除菌级过滤器”专注于最终药物灭菌,没有提到除菌级过滤器重复使用的问题。2008年的版本考虑到了美国之外生物技术产品和医药产品中除菌级过滤器的应用,提到:“单一批次生产之后的除菌过滤器应该进行常规的报废处理。”但这个版本也详细阐明了FDA对于重复使用的无菌工艺指南:“然而,在重复使用能被评估的情形下,除菌过滤器的验证应当包括完整性试、细菌挑战实验和清洁过程并结合将处理的最大数量批次。”
作为两个版本PDA文件的合作者,作者支持PDA的建议,对经过一定限度重复使用和清洁环节的过滤器进行细菌挑战试验。
FDA的警告
已经加强了在GMP下重复使用过滤器的指导。一封由FDA发往无菌医药生产工厂的警告信如是说:“用于多种不同注射剂产品剂型和批次的过滤器未能对扩大的重复使用期进行验证,冲洗验证数据不够充分支持前批产品组分残留已被去除……你们的反应(对483号文件)未能保证产品(节录)不包含源自前批使用同一过滤器的不同产品的不可接受的残留。检查发现支持前批产品残留已被去除的冲洗验证数据不够充分。我们担心交叉污染的可能性。”另一封由FDA发往无菌眼药生产厂的警告信如下:“根据确认的检查报告你们重复使用除菌过滤器,且只要过滤器达到制造商的完整性检验标准(节录)或在你们内控规定的50次最大使用次数内就会长期重复使用。我们关心的是重复使用和高压灭菌50次后你们过滤器的有效性。我们知道每批次生产前后,你们使用(节录)方法对除菌过滤器进行完整性试验。然而,为了评估50次的重复使用,我们见到需要用产品对初次使用的和50次重复使用后的过滤器进行的细菌截留验证研究。还有,我们并不清楚你们是否用产品对过滤器进行了过滤器溶出物和析出物试验。如果你们有这个数据,请提供给我们;若无,请告诉我们何时能够提供。”
一位FDA的评论家公开声明:除菌级过滤器的重复使用“不鼓励用于无菌医药产品”,同时承认除菌级过滤器的重复使用“对API产品很普遍”。该评论家建议:“微生物的截留验证应该结合多次过滤和灭菌过程”,还有“微生物截留验证、多批灭菌/过滤循环后过滤器完整性检测、API/产品无菌性应相关联。”2008年10月的483观察报告中指出:FDA现在要求在批次记录中注明除菌过滤器在之前什么时候重复使用过。
过滤器重复使用验证
根据PDA和FDA所推荐的内容,重复使用的过滤器之无菌过滤验证需要一个多步细菌截留研究计划。PDA的26号技术报告和FDA的无菌工艺指南现要求对产品或者工艺流体进行初步的生物负载研究,来决定用产品过滤器膜片,使用标准细菌挑战生物——缺陷假单胞菌(ATCC19146)或者一种生物负载种类在最恶劣的产品和工艺条件下进行细菌截留研究的适合性。这些细菌挑战试验应该用多批过滤膜片(一般三批),“并且用于细菌截留验证研究的三批膜片中,至少有一张其预研究或使用前物理完整性试验值在或者接近过滤器厂商的试验参数限值”。
此类研究将充分证明在一次性使用时滤膜无菌处理药品或者工艺流体的能力。然而,该研究将不能预测经过多次清洁、重复灭菌和重复使用循环后的过滤器性能。对生产用过滤器应进行细菌挑战试验,在真实的、或首选,最恶劣的工艺条件下进行,并且结合多次清洁、冲洗、重复灭菌和重复使用循环的整个过程。多批次重复使用过程,不论无批次间冲洗还是批次间用溶剂冲洗,经常可以缩小和模拟至实验室规模。然而,更复杂的重复使用过程比如那些需要多次清洗和重复灭菌循环,或者周期烘干的,很难在实验室条件下用膜片、囊式滤器或滤芯模拟的。这时,较好的办法就是在单批次膜片挑战试验基础上,用经过了实际使用、清洗、重复灭菌和重复使用过程的滤芯进行一系列的细菌挑战试验。通常生产滤芯适合于此第二阶段验证,因为“最坏的膜”已在最初的膜片研究时确认,对用过的生产滤芯的实验可用以确认工艺相容性及衡量滤芯细菌截留特性的保持。这种检测是惟一的方法来检测过滤器除菌性能是否受到多次重复使用工艺循环条件的影响。接下来的案例会证明完整性试验可能不够充分。
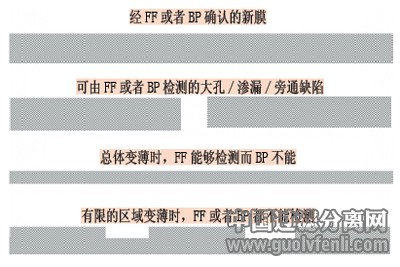
图2 各种可由或不可由前进流试验或者泡点类完整性试验检测的重复使用过程中膜损坏。
重复使用的风险
这里有一个案例研究强调了没有进行全重复使用工艺规模的验证就重复使用除菌级过滤器的一个风险事例。由于保密的原因,此处不涉及特定的工艺细节。某医药公司使用大面积的、打褶的除菌级膜式滤芯,用来过滤大批量无菌API抗生素及其溶剂,而所选择的滤膜和此溶剂有限兼容。这个局限曾被认为是可接受和无关紧要的。每次使用之后,过滤器先用水冲洗,然后碱液清洗。碱液再用水从过滤器上冲洗掉(去除程度未经确认或验证)。过滤器接着进行重复使用批次间的SIP。过滤器在每批次使用前后都进行完整性试验,而且在特定的最大重复使用次数内一直通过了其推荐的检测限值。每个过滤后批次的无菌检测都是常规的,并且没有产品的非无菌报告。为了增加过滤器完整性试验和批式无菌试验的信任度,药物生产厂商对已达到规定最大重复使用寿命的过滤器进行了细菌挑战试验。在ASTM测定液体过滤膜式过滤器细菌截留率的标准检测方法的挑战试验条件下,证实了过滤器的细菌穿透。表明该使用过的过滤器已不再符合除菌级过滤器的定义(亦即当挑战水平>107cfμ/cm2有效过滤面积时,缺陷假单胞菌的截留率为100%)。
重复使用的过滤器细菌截留试验失败了,尽管事实上该过滤器通过了完整性测试,而该测试关联于过滤器厂商在相似的挑战试验条件下对挑战前未经使用的过滤器进行的100%的细菌截留。该研究得到了几个重要的结果:首先,重复使用的过滤器仍然显示出高度的细菌截留能力,但是不再能符合声称的100%缺陷假单胞菌截留率,也不再能符合法规的除菌级过滤器定义。第二,产品中受控的低生物负载,结合稍有降低但仍很明显的过滤器截留特性,能够有效阻止工艺过程中可检测的细菌穿透,这一点已由成功的菌检及不存在非无菌的产品所证明。第三,使用标准的过滤器完整性测试不能检测出重复使用过程中的过滤器损坏。对那些相信过滤器完整性试验可以检测出任何大孔、渗漏、或影响除菌过滤性能的缺陷的人,这一结果看来是个反证。这些试验如前进流或泡点试验的关联性基于对新过滤器及那些有在过滤器生产、使用和安装的过程中造成的实际损伤或缺陷的滤膜或滤芯进行的细菌挑战试验。过滤器厂商的细菌截留验证和完整性试验关联性研究所用的过滤器不包括那些由于终端用户重复使用工艺不兼容而损坏的过滤器。这些不兼容性可由未认证、清洗、再次灭菌和重复使用而产生。
完整性试验的局限
过滤膜一般被认为是很多圆柱形毛细管,其中细菌截留仅由对大于膜上最大孔径的细菌的尺寸拦截单独控制。依照这个模型,泡点类试验可以检测膜上超标大孔或者缺陷(亦即大孔,密封旁路)的存在,前进流试验可以提供流量的定量测量已证明没有超标大孔或者缺陷。然而,微孔滤膜对细菌的截留作用不单单是一种由小于细菌的毛细孔而产生的尺寸拦截功能。膜的其他性质也对细菌的截留有贡献,如多孔结构的形状和曲折性、膜的厚度(亦即从上游到下游穿过膜孔的流道长度)、可能发生在细菌和任何足够容纳细菌的大孔壁之间的吸附力等。前进流和泡点试验都不足以检测出这些次要截留因素的改变。
这些条件的不良变化通常不会发生在经适当验证的滤膜制造过程中,完整性试验在检测这些截留变化偏差上的局限没有得到充分的重视。
通过对重复使用的打褶滤芯进行故障分析,我们最终明确了过滤器细菌穿透的根本原因。多次重复使用中的损坏归结于膜的化学降解,使得滤芯的褶型顶端变薄,如图1所示。局部的化学损坏和褶型顶端变薄通常指示过滤器的局部干燥,在蒸汽流过褶型顶端的过程中,那些能够在热蒸汽条件下对滤膜造成化学腐蚀的流体组分会在这些部位富集。
此例中,膜厚度的局部降解归因于在重复使用的SIP重复灭菌阶段热碱的作用。SIP之前残留碱的存在是由于碱清洗剂的冲洗不充分,随后灭菌前滤芯上水的蒸发引起在重复使用的SIP重复灭菌阶段褶型顶端的碱浓度增加。褶型顶端残留浓碱上的蒸汽使温度的逐渐升高从而加速了上述部位膜表面的化学降解。局部区域膜厚度的降低足以能够让细菌穿透。然而,由于这种损坏并没有穿过整个膜(没有洞或者过大的孔),变薄的区域局限在褶型顶端很小的面积上,无论泡点试验还是前进流试验没有超过其各自的通过/失败限值。
如图2所示,泡点试验只能检测全厚度的大孔缺陷。前进流扩散流类试验,原则上与膜厚度相关,但是不能检测出此种情况下仅存在于褶型顶端有限的变薄区域。由于变薄区域并未提高前进流量到超过试验限值,所以不会被检测到。
结论
法规并不鼓励除菌过滤器的重复使用,特别是无菌的医药产品。经过评估,除菌过滤器也许可以在某些情况下重复使用,但是重复使用必须得到验证,即不会降低过滤器除菌性能或者滤出物质量。除了基础的灭菌验证研究之外,依据PDA26号技术报告和FDA的无菌工艺指南的推荐内容,多次重复使用除菌过滤器的验证应该包括对经最大特定数目的清洁,重复灭菌,烘干和重复使用后过滤器的彻底检验。彻底检验的内容包括细菌挑战试验和过滤器完整性试验。这些试验应测试过滤器对工艺流体和重复使用条件的化学兼容性、在重复使用条件下完整性试验的有效性及冲洗滤出液的化学分析,以确认任何细菌析出物,产品或者清洗剂残留物。
没有适当地对使用过的滤芯的细菌挑战验证试验,不能仅靠完整性试验来说明重复使用的过滤器的除菌性能。最后,在设计和确认无菌过滤工艺时,使用者应该认真考虑风险水平以及除菌过滤器的满意使用中所含的验证费用,并与过滤器重复使用得到的表面经济效益相比较。
清洗滤芯
清洗过程是比简单地冲洗和重复使用更为复杂的问题。清洗过程指的是去除源自整个系统包括所有的管道、储罐、阀门和过滤器的前批次残留物。最终的冲洗步骤完成之后,注射用水(WFI)冲洗(或者纯溶剂冲洗)须由验证过的分析方法证明该系统达到一个预设的洁净度。如果使用了在线清洗材料,也必须将其完全地冲洗掉。记住清洗过程可能会将上游储罐和管路中的物质过滤并留在过滤器中。这种截留物能析出到下一批次产品中。开发和验证一个合适的清洗方法是每一位使用者的责任。使用者应该就将用到的清洗方法的合适性向过滤器厂商咨询。针对液体过滤器的CIP,还有几点需要考虑:
某些过程流体可以为细菌提供养分,将导致作为截留细菌降解副产物的过滤器上热原细菌内毒素。内毒素的含量必须保持在低水平。因此,可生长细菌流体从一个产品周期传递到下一个周期是有问题的。
过滤器一旦有任何可察觉的堵塞时(亦即,低流速或者高压差),流体不易流过堵塞的滤孔,因此阻碍了清洗溶液到达该堵塞点。
清洗流体优先流过较干净的流道,而“较脏”的堵塞路段事实上没有受到作用。
一些产品残留和清洗剂组分总会或多或少存在于过滤器内。对于残留的可滤去物质,任何清洗过程都需要有一个可以接受的残留可析出物限度,而该物质的检测也需要一个既定的经验证的方法。
制药领域过滤器的清洗和重复使用的困难大小将基于下述因素的大小:
进料液细菌生物负载的水平;
产品支持细菌生长的活性;
过滤器堵塞状况;
产品、产品组分或者副产物的生物活性;
产品、产品组分或者副产物的耐溶解性;
产品、产品组分或者副产物对过滤器材料的粘着性;
选择一种产品适合的无菌或者抑菌的过滤器存储方法的难度。
这些风险正好阐述了人们使用一次性过滤器的原因。通常,考虑到这些风险时,药物产品和过程流体要明显比过滤器更加贵重,产品质量和患者安全的风险太大以致过滤器的重复使用失去了吸引力。
 加利利.上海加利利阀门.加利利阀门.上海加利利
加利利.上海加利利阀门.加利利阀门.上海加利利 匠奇
匠奇 固安宇晨滤清器厂
固安宇晨滤清器厂 深圳威马森过滤技术有限公司
深圳威马森过滤技术有限公司 固安县顺峰滤清器厂
固安县顺峰滤清器厂 江苏阜升环保滤袋有限公司
江苏阜升环保滤袋有限公司 天津市鑫东水处理设备有限公司
天津市鑫东水处理设备有限公司 深圳市创生源
深圳市创生源 东莞龙田过滤设备
东莞龙田过滤设备 鸿淳环保垃圾渗滤液处理技术服务
鸿淳环保垃圾渗滤液处理技术服务 河北廊坊油分滤芯1604039381
河北廊坊油分滤芯1604039381 雷尼镍催化剂过滤器
雷尼镍催化剂过滤器 专业供应22111975 32128883油分
专业供应22111975 32128883油分 液压油过滤芯纸,滤芯纸,液压油滤芯
液压油过滤芯纸,滤芯纸,液压油滤芯 HC2286FKS12H50YT电厂液压油滤芯厂家直销
HC2286FKS12H50YT电厂液压油滤芯厂家直销 NDS重型卧式中开多级离心泵
NDS重型卧式中开多级离心泵 除尘滤芯P191920-016-436空气滤芯
除尘滤芯P191920-016-436空气滤芯 久骥过滤器封盒机,滤芯器封盒机,优质半自动封盒机专业品牌,值得信赖!yTbKH
久骥过滤器封盒机,滤芯器封盒机,优质半自动封盒机专业品牌,值得信赖!yTbKH 树脂过滤芯临汾 涂装过滤芯
树脂过滤芯临汾 涂装过滤芯 大量求购二手压滤机.过滤机s3k
大量求购二手压滤机.过滤机s3k 高价求购二手200平方冬化过滤机
高价求购二手200平方冬化过滤机 过滤袋广州 液体过滤袋深圳 过滤液体袋东莞YKmAG
过滤袋广州 液体过滤袋深圳 过滤液体袋东莞YKmAG 不锈钢过滤器生产
不锈钢过滤器生产 求购五芯精密过滤器
求购五芯精密过滤器 成都怡尚科技四川传递窗,风淋室,过滤器,净化工程供应YKgad
成都怡尚科技四川传递窗,风淋室,过滤器,净化工程供应YKgad






